





Recognise the signs, identify the disease
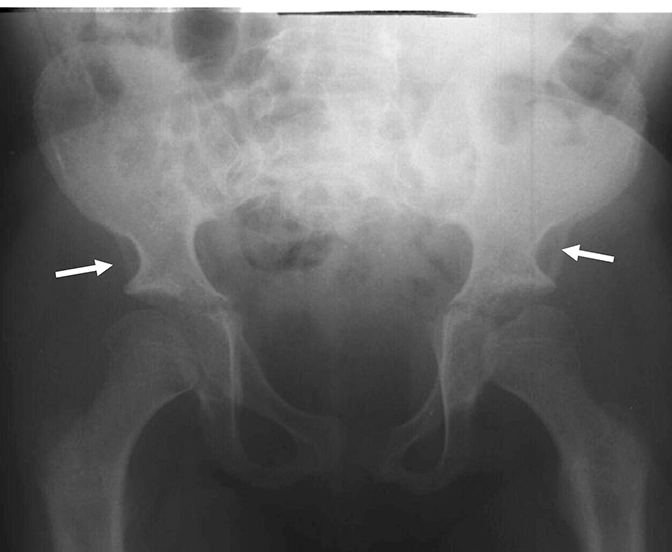
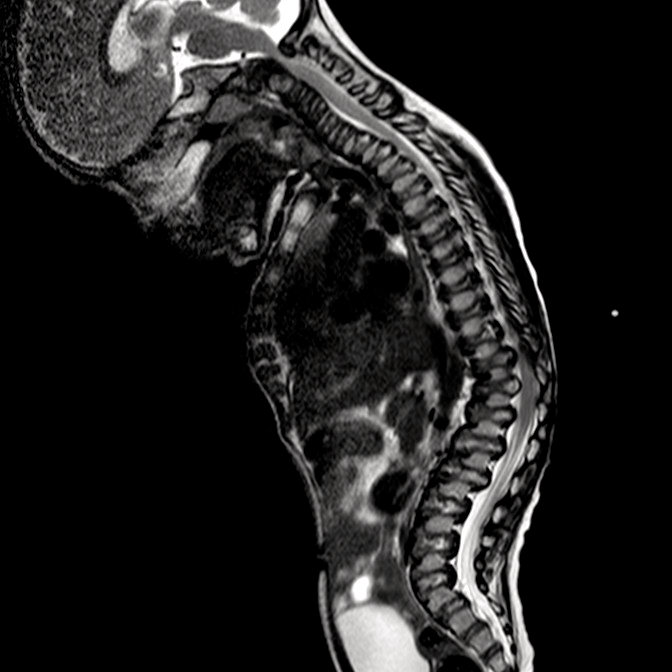
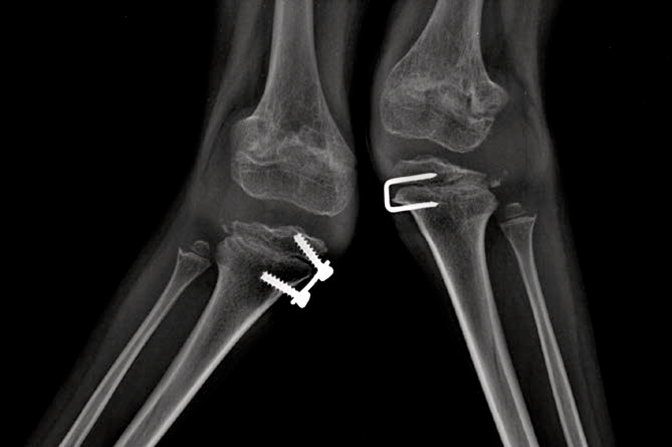
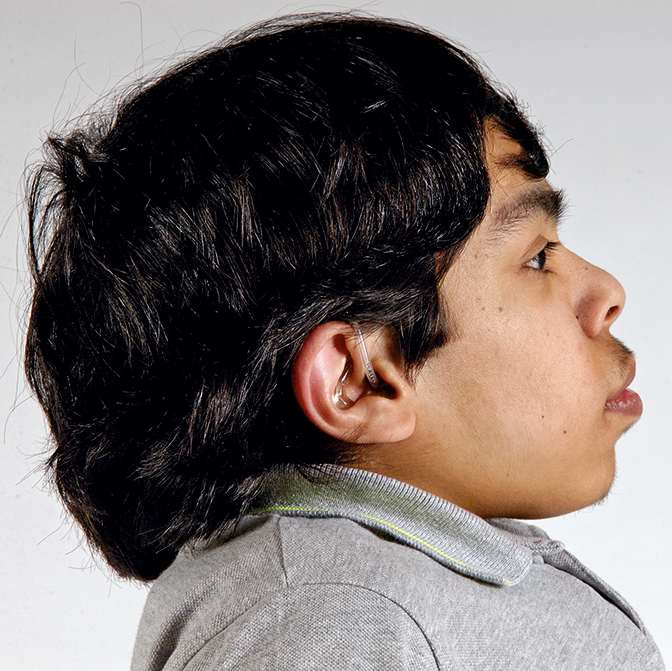
Mucopolysaccharidosis (MPS) disorders are a group of progressive, genetically inherited, serious disorders that affect multiple body systems and functions as a result of heterogeneously expressed enzyme deficiencies.1-3 Variability in presentation and disease progression — as well as perceived disease rarity in broad clinical practice — result in an under recognition of MPS in specialty practices, leading to delayed diagnosis and potentially devastating consequences.4,5
Optimise long-term patient outcomes and help initiate early treatment by recognising the signs of MPS and referring suspected patients to a geneticist or metabolic centre immediately. Referral to a metabolic centre is a critical first step in the differential diagnosis and early initiation of treatment.4
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}