


Age at diagnosis: 3 years
Time to diagnosis: <1 yeartrong>
Key diagnostic symptom: Heart murmur
Diagnosing doctor: Paediatric cardiology
Patient with MPS VI, ages 0 to 17
Description1:
Summary:
Early diagnosis of MPS VI enabled early intervention and management, which are associated with improved clinical outcomes.2
Classic MPS symptoms – short stature, recurrent otitis media, skeletal and rheumatological manifestations – with accompanying cardiac involvement should prompt referral to a geneticist or metabolic centre.2
Enzyme replacement therapy (ERT) initiated





Description1:
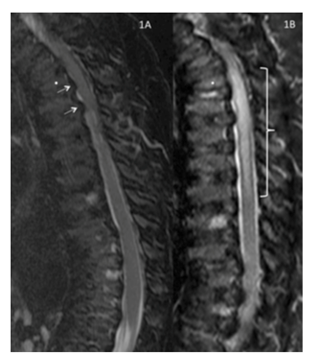
Sagittal T2/short T1 inversion recovery – weighted image of the thoracic and lumbar spine obtained 7 months preoperatively.
Reproduced with permission from Drummond, Can J Anesth, 2015.
Summary1:
In this illustrative case, postoperative spinal cord injury was disproportionate to the degree of spinal stenosis. This case is an example of the exaggerated vulnerability of people with Morquio A to spinal cord compression and damage.
Anaesthesiologists should consider subclinical spinal stenosis with spinal column joint laxity adding to the risk of compression.2
Case history1
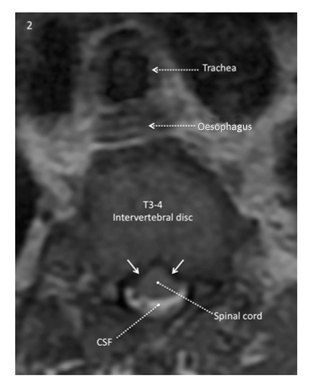
The axial T2-weighted image at the level of the T3-4 intervertebral disk obtained 7 months preoperatively shows cerebrospinal fluid (white) around the posterior portion of the spinal cord but not anterior. The anterior surface of the cord has a “tented” rather than a smooth convex configuration, reflecting bilateral ventrolateral pressure on the anterior surface of the cord (solid white arrow).
Reproduced with permission from Drummond, Can J Anesth, 2015.
Based on a case study provided by Dr Zakharchuk
Description1:
Case history1
Summary1
Early diagnosis is key to initiating ERT, when available, and providing opportunities to improve patient outcomes.4-7,b As this case demonstrates, ERT has the potential to improve key clinical parameters, such as endurance and respiratory measures, which may be critical to patient quality of life, maintenance of ambulation, and activities of daily living.8,9
Age at diagnosis: 12 years
Time to diagnosis: 11 years
Key diagnostic symptom: Motor and speech delays
Diagnosing doctor: Geneticist
Onset of neurological symptoms at 1 year of age, followed by rapid progression of developmental delay, leads to diagnosis of MPS IIIA in 2 siblings1
Description1:
Summary1:
As this case demonstrates, signs and symptoms can progress rapidly, particularly with neurological deterioration and worsening developmental delay in patients with MPS IIIA. For families with multiple children demonstrating any features indicative of MPS, the index of suspicion should be especially high.1
While continual monitoring and evaluation are critical, early intervention through direct referral to a geneticist and/or metabolic centre, rather than metabolic screening using urinary GAGs, is recommended to expedite diagnosis.1
Case history1
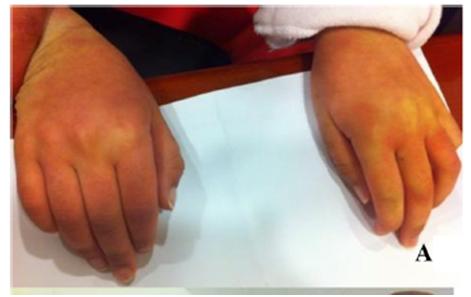
Examination reveals clear skin discolouration in patient with MPS IIIA.1
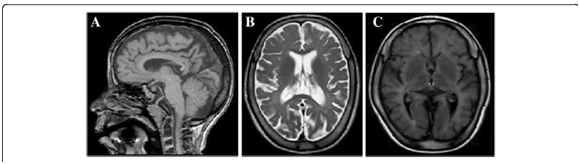
MRI revealed diffuse hypomyelination, thinning of the corpus callosum, and moderate cerebral atrophy, symptoms that developed over time.1
References: 1. Data on file. Biomarin Pharmaceutical. 2. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1-15. doi:10.1002/ajmg.a.36833. 3. Giugliani R, Lampe C, Guffon N, et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome)–10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am J Med Genet A. 2014;164A(8):1953-1964. doi:10.1002/ajmg.a.36584.
References: 1. Drummond JC, Krane EJ, Tomatsu S, Theroux MC, Lee RR. Paraplegia after epidural-general anesthesia in a Morquio patient with moderate thoracic spinal stenosis. Can J Anesth. 2015;62(1):45-49. doi:10.1007/s12630-014-0247-1. 2. Spinello CM, Novello LM, Pitino S, et al. Anesthetic management in mucopolysaccharidoses. ISRN Anesthesiol. 2013;2013:1-10. doi:10.1155/2013/791983.
References: 1. Data on file. Biomarin Pharaceutical. 2. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1-15. doi:10.1002/ajmg.a.36833. 3. Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010;5:5. doi:10.1186/1750-1172-5-5. 4. Clarke LA. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology (Oxford). 2011;50(suppl 5):v13-18. doi:10.1093/rheumatology/ker395. 5. Lehman TJA, Miller N, Norquist B, Underhill L, Keutzer J. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41-v48. 6. Morishita K, Petty RE. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v19-v25. doi:10.1093/rheumatology/ker397. 7. Muenzer J. Overview of the mucopolysaccharidoses. Rheumatology. 2011;50:v4-v12. doi:10.1093/rheumatology/ker394. 8. Hendriksz C. Improved diagnostic procedures in attenuated mucopolysaccharidosis. Br J Hosp Med. 2011;72(2):91-95. 9. Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111(2):63-72. doi:10.1016/j.ymgme.2013.11.015.
Reference: 1. Sharkia R, Mahajnah M, Zalan A, Sourlis C, Bauer P, Schöls L. Sanfilippo type A: new clinical manifestations and neuro-imaging findings in patients from the same family in Israel: a case report. J Med Case Rep. 2014;8:78. doi:10.1186/1752-1947-8-78.
